
Обычно при рассмотрении механизма полиэтерификации принимают, что реакционная способность функциональных групп не зависит от длины молекулярной цепи, которой она принадлежит и от вязкости реакционной среды, которая сильно возрастает при поликонденсации (принцип Флори). Рассмотрение этих предложений позволяет при оценке кинетики пользоваться единой константой скорости реакции конденсации и концентрациями функциональных групп [1].
Скорость линейной поликонденсации измеряется скоростью изменения концентрации одной из расходуемых в реакции функциональных групп (Cа или Сb):
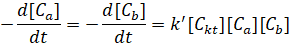 (1)
(1)
где k' – константа скорости. [Ckt] – концентрация катализатора, которую принимают постоянной в течении всего процесса, то:
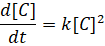 (2)
(2)
где 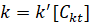 .
.
Катализаторами поликонденсации служат те же соединения, которые катализируют аналогичные реакции монофункциональных веществ. Например, в качестве катализаторов полиэтерификации используют минеральные кислоты, сульфокислоты, кислые соли.
Равновесные процессы характеризуются малыми скоростями (k ~ 10‑3÷10-5 л/моль·с) и сравнительно высокими значениями энергии активации (85÷170 кДж/моль); они могут быть экзо- и эндотермическими. Для большинства случаев неравновесной поликонденсации характерны высокие скорости (k достигает 105 л/моль·с) и низкие значения энергии активации (8÷40 кДж/моль); эти процессы, как правило, экзотермичны.
Средняя степень полимеризации образующегося полимера Рn в отсутствии реакций обратной росту выражается в виде:
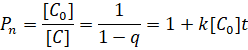 (3)
(3)
В соответствии с этим определением величина Рn для поликонденсационных процессов учитывает вклад всех n-меров, включая вклад молекул мономеров, присутствующих в реакционной среде.
Из уравнения (1) следует, что высокомолекулярные продукты могут быть получены только при степенях превращения близких к 1 (q>0.95). При меньших степенях превращения образуются лишь олигомерные вещества [2].
При линейной поликонденсации предельное значение степени полимеризации должно теоретически бесконечно увеличиваться с ростом глубины превращения.
Достичь степени полимеризации, превышающей 103 очень трудно. Одна из причин этого – трудность достижения строгой стехиометрии реагирующих функциональных групп из-за протекания побочных реакций.
Причиной понижения Рn может быть присутствие монофункциональных веществ, присоединение которых к концу растущей цепи приводит к образованию нереакционноспособных концов. Монофункциональные соединения могут образовываться в реакционной системе в результате побочных реакций, например, декарбоксилирования карбоксильных групп при повышенных температурах. В некоторых случаях небольшие количества монофункциональных соединений специально вводят в реакционную смесь для регулирования молекулярной массы образующихся полимеров.
Другой причиной ограничения растущих цепей при поликонденсации являются реакции циклизации. В отличии от линейной конденсации при циклизации реагируют функциональные группы одной и той же молекулы, приводящие к образованию циклов, не способных к дальнейшему росту цепей.
Основными факторами ограничения молекулярной массы при линейной равновесной поликонденсации являются обратимость основной реакции, а также деструкция образовавшихся макромолекул в результате их побочных реакций с низкомолекулярными веществами.
В отсутствии побочных реакций предельно достижимая степень полимеризации при равновесной поликонденсации определяется термодинамическими факторами.
В большинстве случаев молекулярная масса поликонденсационных полимеров определяется не термодинамическими, а кинетическими факторами и стехиометрией состава мономерной смеси.
Образование активных центров на поверхности частиц катализатора.
Характерным свойством оксидов переходных металлов является способность иона металла достаточно легко переходить из одного состояния окисления в другое [3, 4]. У оксидов переходных металлов, как в объеме, так и на поверхности кристалла имеется или недостаток, или избыток кислорода по сравнению со стехиометрическим составом [4]. Избыточные ионы О2- достраивают кристаллическую решетку с образованием катионных вакансий, а катионы вблизи таких вакансий переходят в высшее состояние окисления. При дефиците кислорода образуются анионные вакансии, а ионы металла вблизи таких дефектов находятся в низшем окислительном состоянии. Ионы металлов, катионные и анионные вакансии на поверхности кристалла могут выступать в качестве активных центров в катализе [4]. Катион кристаллической решетки оксида можно рассматривать как центральный ион в комплексном соединении переходного металла. Поскольку поверхностные ионы металла координированы не полностью, то свободные места в их координационной сфере занимают молекулы реагента, вступающие в химическую связь с катализатором. Таким путем образуются поверхностные промежуточные соединения, участвующие в катализе. Число дефектов кристаллической решетки оксида, степень окисления иона металла, его координационное число можно изменять путем введения в объем или на поверхность катализатора оксидов других металлов. Смешанные катализаторы обладают значительно более высокой каталитической активностью по сравнению с индивидуальными оксидами [3].
Высокая каталитическая активность переходных металлов связана с наличием «свободных» d-АО поверхностных атомов. Последние участвуют в образовании химических связей с молекулами реагентов, находящихся на поверхности. При этом одна молекула реагента может образовать связь с одним, двумя и более атомами поверхности. Таким образом, один или группа атомов на грани кристалла может рассматриваться как активный центр катализатора.
а) имеются катионные вакансии в решетке, но нет анионных вакансий (FeO, CoO, NiO, Cu2O, Cul);
б) имеются анионные вакансии в решетке, но нет катионных вакансий (NaCl, KCl, Kbr);
в) катионы занимают промежуточные положения в решетке (ZnO, CdO);
г) анионы занимают промежуточные положения в решетке (UO2);
Существуют следующие неэмпирические методы и модели для изучения адсорбции реагентов на поверхности частицы катализатора: кластерная модель, модель двупериодической пластины, модель трехпериодической пластины [5].
Полиэтерификация относится к типу поликонденсационного процесса, включающего акты присоединения-отщепления. В кислотных реагентах атомы углерода карбонильной группы имеют частичный положительный заряд в следствии наличия кратной связи с электроотрицательным атомом кислорода и представляют собой электронодефицитные центры, способные вступать во взаимодействие с нуклеофилом. При этом промежуточное соединение представляет собой продукт присоединения атакующего реагента к поляризованным атомам карбонильной группы.
Образование переходного состояния является медленной стадией процесса. Степень поляризации карбонильной группы в переходном состоянии зависит от природы нуклеофильного агента. Установлено, что лимитирующей стадией полиэтерификации является нуклеофильная атака карбонильной группы.
Основное действие катализатора проявляется в активации реагирующих молекул, т.е. в усилении их нуклеофильных или электрофильных свойств. Это приводит к увеличению скорости реакции и снижению энергии активации в следствии изменения реакционного пути.
Процесс полиэтерификации лимитируется процессом диффузии через инертный слой жидкой фазы окружающей частицу катализатора и скорость определяется скоростью переноса реагентов с поверхности слоя жидкости на поверхность катализатора.
Моделирование процесса диффузии реагентов через жидкий слой катализатора.
Концентрации веществ в ядре потока и на поверхности слоя жидкости частицы катализатора в следствии интенсивного перемешивания равны. На поверхности частицы катализатора существует диффузионный слой, толщиной , где основной механизм переноса реагентов – молекулярная диффузия. Изменение концентрации исходных реагентов и продуктов реакции можно представить графиками на рис. 1.
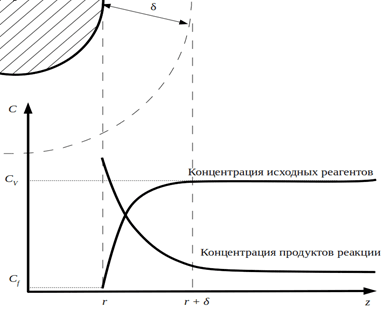
Рис. 1. Изменение концентрации исходных реагентов и продуктов реакции в слое жидкости и на поверхности частицы катализатора
Концентрации реагентов на поверхности частицы катализатора (Cf) для процессов, протекающих в диффузионной области близка к нулю, концентрации реагентов в ядре потока (CV) при периодическом ведении процесса будут меняться во времени.
Так как размеры частиц катализатора в наноструктурированной форме малы пренебрегать кривизной поверхности не можем, следовательно необходимо рассматривать молекулярный перенос вещества в слое жидкости к поверхности катализатора в сферической системе координат.
При постоянном значении коэффициента молекулярной диффузии (D), поток вещества к частице катализатора в таких условиях можно описать следующим уравнением:
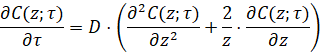 (4)
(4)
Начальное условие уравнения (4) соответствует равенству концентрации вещества в переходной зоне и в ядре потока:
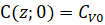 (5)
(5)
Граничные условия соответствуют условиям первого рода (задана концентрация на поверхности и границе диффузионного слоя):
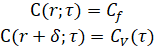 (6)
(6)
При этом поток реагентов к поверхности частицы катализатора рассчитывается по зависимости:
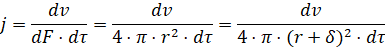 (7)
(7)
Изменение количества реагентов в ядре потока определятся по уравнению:
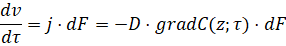 (8)
(8)
Величина коэффициента D в уравнении (4), соответствует коэффициенту молекулярной диффузии только на границе z=r, поэтому для обеспечения необходимой точности расчета величину градиента концентрации следует брать при данной координате:
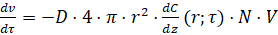 (9)
(9)
В уравнении (9) производная концентрации по радиусу частицы катализатора при является движущей силой процесса диффузии, аналогично  при диффузии описываемой пленочной моделью. Для определения величины движущей силы диффузионного переноса необходимо решить уравнение (4) при условиях (5) – (6).
при диффузии описываемой пленочной моделью. Для определения величины движущей силы диффузионного переноса необходимо решить уравнение (4) при условиях (5) – (6).
Выделение переходной зоны шириной позволяет учесть вклад гидродинамических процессов (при более активном гидродинамическом режиме значение толщины переходного слоя будет уменьшаться).
В более сложных, либо требующих большей точности расчета случаях можно применять зависимость вида С(δ+r;τ)=Kc⋅CV, где коэффициент Kc<1 при CV>Cf и Kc>1 при CV<Cf учитывает отличие концентрации на границе диффузионной области и в ядре потока (рис. 2).
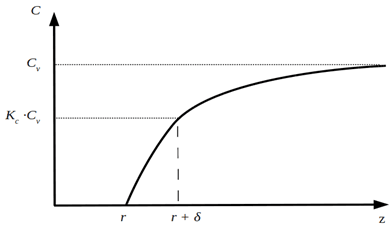
Рис. 2 Изменение концентрации реагентов в диффузионной области и на поверхности частицы катализатора
Толщина диффузионного слоя (δ) определяется зоной молекулярного переноса реагентов при постоянном коэффициенте диффузии.
Для использования кинетической зависимости (9) необходимо экспериментально определить величины δ, D, r, KC, и движущую силу процесса решив уравнение (4).
Выводы
Предложена физическая модель процессов полиэтерификации при производстве органического пленкообразующего вещества с использованием катализатора в наноструктурированной форме. Получены уравнения, описывающие процесс диффузии реагентов к поверхности частиц катализатора в сферической системе координат. Разработана математическая модель процесса полиэтерификации, позволяющая получить значения степени превращения сырья в любой момент времени.
.png&w=640&q=75)