
Краниосиностоз представляет собой врожденное заболевание, характеризующееся преждевременным сращиванием черепных швов у новорожденных. Это может привести к различным последствиям, таким как деформации черепа, увеличение давления внутри черепа, задержка развития мозга и неврологические проблемы. Распространенность краниосиностоза оценивается в 1 случай на 1100–2500 новорождённых.
Возможные причины заболевания:
На данный момент этиология данного заболевания не выяснена до конца. Одним из главных считается эмбриональный фактор: это болезни обмена веществ, например заболевания щитовидной железы у матери, дефект формирования мезенхимальной капсулы, внутриматочная компрессия черепа [1, с. 2-3]. Так же краниосиностоз возможен в случае генетических нарушений. В частности, это аномалия генов FGFR (включающих в себя FGFR1, FGFR2, FGFR3) рецепторов фактора роста фибробластов, которые влияют на дифференцировку клеток нервного гребня [2, с. 1-2]. Предположено наличие связи между мутациями в MSX2, TWIST и нарушениями развития черепно-лицевого скелета, при этом появляется сбой роста хрящевых клеток в клиновидной кости, из-за чего появляются нарушения в формировании окружающих костей у ребенка.
Классификация краниосиностозов:
Краниосиностоз подразделяют на два вида:
- Несиндромальный – изолированная форма, которая возникает самостоятельно и не имеет сопутствующих заболеваний, чаще всего встречается. Составляет до 70% всех краниосиностозов, его частота 1:1000-1:2500 новорожденных. Несиндромальный КС чаще выявляются у мальчиков, что может так же свидетельствовать о гормональном влиянии андрогенов на развитие заболевания [3, с. 922-927].
- Синдромальный – патология сочетается с другими врожденными пороками такими, как Синдром Аперта, синдром Пфайффера, синдром Крузона и синдром Антли-Бикслера, ведь они связаны с мутациями в семействе FGFR (особенно в семействе FGFR2) [2, с. 1-2]. Составляют 15–30% от общего количества заболеваний.
В зависимости от количества заросших черепных швов выделяют:
- Моносиностоз – характеризуется поражением только 1 шва. В случае с коронарным и лямбдовидным швом зарастание может быть одно- или двухсторонним, наиболее распространенная форма из всех видов.
- Полисиностоз – в патологический процесс втягиваются 2–3 шва.
- Пансиностоз – сращение всех костных швов черепа ребенка, встречается наиболее редко.
Краниосиностоз подразделяют так же в зависимости от самого шва:
- Скафоцефалия (сагиттальный шов) – череп приобретает удлиненный, суженный вид. Является самым распространенным, встречается в 40% случаев. Чаще всего проявляется у мужчин в связи с большим размером головы у плода на III триместре беременности, из-за чего происходит ограничение в движении и возможная компрессия черепа ребенка [1, с. 2-3].
- Лобная и затылочная плагиоцефалия (коронарный шов и лямбовидный шов соотвественно, с одной стороны) – в случае лобной имеет одностороннее уплощение лобной кости, смещение верх глазницы на пораженной стороне, девиация носовой перегородки, а при зарастании лямбовидного шва появляется одностороннее уплощение затылочной кости [4, с. 23-25]. В целом, составляет около 20–30 процентов случаев. Чаще всего встречается у женского пола, причем с правой стороны [6, с. 913-921]. В следствии могут проявляться нарушения прикуса, одностороннее косоглазие.
- Тригоноцефалия (метопический) – лоб становится клиновидной формы или череп приобретает килевидную форму, встречается около 10% случаев. Заболевание наследуется в 2–8 % случаев. Так же, как и в случае скафоцефалии чаще встречается у мужчин, может быть связана с аномалиями хромосом 3, 9, 11 и сочетаться с аномалиями ЦНС [5, с. 55-59].
Диагностика краниосиностозов:
У беременных женщин краниосиностоз плода возможно выявить при ультразвуковом исследовании, 3D УЗ-сканировании с 28-й по 40-ю неделю беременности (III триместр беременности).
Возможные патологии:
- Закрытый родничок у новорожденного;
- Череп удлинен, присутствует выступ в области лба;
- Наблюдается наклонный лоб и затылок;
- Асимметрия положения глаз;
- Деформация головы, наличие костного выступа у большого родничка и другие.
При выявлении данных патологий рекомендуется кесарево сечение, для минимализации травм головы ребенка. Но чаще всего данный диагноз ставят после рождения ребенка, обращая внимание на деформацию формы головы и сращивание швов.
Лечение краниосиностозов:
Лечат краниосиностоз только операцией – чем раньше ее сделают, тем лучше. Оптимальный возраст до 6 месяцев, в идеале – до 3 месяцев. В этом возрасте проводится эндоскопическая операция. Хорошие результаты могут быть достигнуты до 9 месяцев. У детей после года проводится обычная реконструктивная операция, но у них деформация головы хуже поддается коррекции, уже могут быть неврологические осложнения.
Существуют три вида операции:
- Малоинвазивная краниотомия. Проводят при несиндромальном краниосиностозе в возрасте до 6 месяцев. Хирург делает разрезы на коже головы и с помощью эндоскопа следит за правильным выполнением операции: он удаляет кость вместе с заращенным швом, притом делая клиновидные разрезы для обеспечения мозга свободным пространством. После операции необходимо ношение специального шлема для правильной реконструкции формы черепа.
- Реконструктивные операции. Проводят при простых и сложных несиндромальных и синдромальных краниосиностозах. Оптимальный возраст – 6–9 месяцев, но можно делать и детям старше. После виртуального планирования хирург делает широкие разряды костей, выполняет ремоделирование костей свода и основания черепа, реконструкции областей черепа с использованием разных методов остеосинтеза (с помощью титановых винтов и пластин, биодеградируемых элементов фиксации).
- Комбинированные методы лечения - краниотомии или небольшие реконструкции сочетаются с другими методами, например дистракцией. Показаны в случае синдромальных и несиндромальных сложных краниосиностозов в возрасте 3–6 месяцев. Участок черепа разрезают, устанавливают дистракционный аппарат, с помощью которого и будут постепенно удалять кости друг от друга.
Лечение краниосиностозов, до/после (автор фотографий: Павел Голованев):
Тригоноцефалия
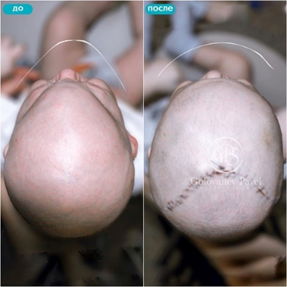
Фото 1
Ребенку 4 месяца, была проведена операция – краниопластика, в ходе которой были установлены биорезорбируемые минипластины. Результат через 7 дней после операции.
Плагиоцефалия
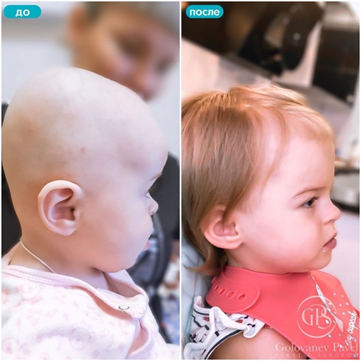
Фото 2
Возраст девочки 8 месяцев, была проведена краниопластика – реконструкция черепа. Результат через 1 год после операции.
Скафоцефалия
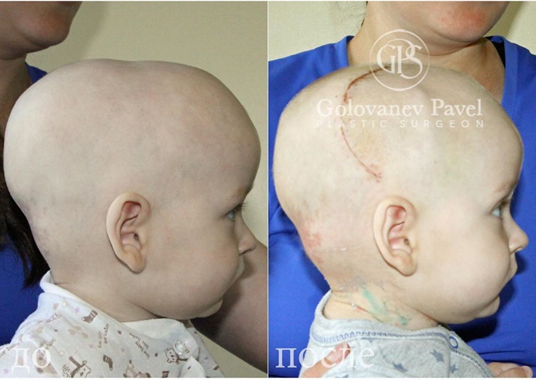
Фото 3
Ребенок 7 месяцев. Была проведена краниопластика с фиксацией с помощью биорезорбируемых минипластин.
Заключение
Краниосиностоз представляет заболевание, при котором из-за закрытия швов черепа, мозг ребенка и другие органы не могут правильно сформироваться, что может привести к различным осложнениям, неврологическим серьезным последствиям. Необходимо, как можно раньше диагностировать краниосиностоз, ведь для благоприятного развития ребенка, необходимо оперативное вмешательство до 6 месяцев. Своевременное лечение может значительно улучшить прогноз и качество жизни ребенка, предотвратить дальнейшие осложнения и обеспечить нормальное развитие когнитивных и физических функций. Важно не только оперативное вмешательство, но и последующее наблюдение и поддержка развития ребенка с помощью специализированных программ реабилитации.
.png&w=640&q=75)