
Введение
На фоне роста социально значимых заболеваний все более важным становится ограничение бюджетных расходов на здравоохранение, в связи с чем, значительную роль в мировой экономике играет разработка генериков, которые являются непатентованными копиями оригинальных лекарственных препаратов [2, с. 23-26]. Создание и регистрация таких препаратов снижает перерасход средств, приблизительно оцениваемый в 150–800 млн долларов на каждый оригинальный препарат, что в последующем регулировать цены на лекарства, делая их более доступными. Перспектива получения того же лекарства с меньшими затратами заманчива, а спрос на непатентованные лекарства растет во всем мире, и фармацевтические компании осознают этот огромный потенциал. В настоящее время, доля воспроизведенных или генерических препаратов на фармацевтических рынках различных стран составляет значительную величину, так в Российской Федерации она составляет 78–95%, в странах Европы – 35–55%, в странах Востока – 30–70%, а в США – 12–15% [1, с. 87-94; 2, с. 23-26; 8, с. 260-267; 11, с. 338-349].
Однако, несмотря на востребованность генерические лекарственные средства остаются постоянным предметом жарких дискуссий, что связано с ускоренным процессом разработки, клинических испытаний и одобрения их использования человеком, а также периодическими сообщениями о побочных эффектах и смертях, связанных с их применением [6, с. 27-30; 7, с. 5-13; 8, с. 260-267]. Очевидно, что важнейшими факторами, влияющими на качество непатентованных лекарств, является соблюдение международных стандартов (Good Manufacturing Practice, GMP) в рамках соответствующих параметров для обеспечения желаемого качества генерика [2, с. 23-26; 4; 5, с. 1335-1355; 6, с. 27-30; 7, с. 5-13; 8, с. 260-267; 9]. В настоящей работе рассмотрены основные принципы и этапы разработки, а также пути регулирования качества генерических препаратов в мировой фармацевтической практике.
1. Основные характеристики генерических лекарственных средств
Термин «генерик» («джнерик») появился в 70-е годы прошлого столетия, что было обусловлено объединением препаратов–аналогов под одно родовое (генерическое) имя, родоначальником которого считалось оригинальное лекарственное средство, которое продавалось под специальным торговым наименованием [3, с. 5-11; 8, с. 260-267]. Смысл этого правила заключался в том, что оно облегчало распознавание оригинального препарата среди аналогов. В настоящее время генерики часто имеют собственные названия, что не позволяет отличать их от оригинала. Основными признаками генерика являются [1, с. 87-94; 2, с. 23-26; 3, с. 5-11; 4; 5, с. 1335-1355]:
- относительно низкая стоимость;
- отсутствие патентной защиты;
- назначение и продажа под непатентованным наименованием (МНН);
- почти полное соответствие оригинальному продукту по составу (за исключением вспомогательных веществ);
- соответствие фармакопейным требованиям;
- производство в условиях GMP (надлежащая производственная практика).
Согласно международному стандарту генерик, или воспроизведенное лекарственное средство – это лекарственный продукт с доказанной фармацевтической, биологической и терапевтической эквивалентностью с оригиналом [1, с. 87-94].
Фармацевтическая эквивалентность – это содержание одних и тех же активных субстанций в одинаковом количестве и лекарственной форме, соответствующих требованиям одних и тех же или сходных стандартов [5, с. 1335-1355]. Фармацевтически альтернативными являются препараты, включающие одинаковый действующий компонент, содержащийся в виде различных химических комплексов или различных дозах [6, с. 27-30].
Биологическая эквивалентность – это скорость и степень всасывания оригинала и генерика в одинаковых дозах по концентрации в жидкостях и тканях организма [9].
Терапевтическая эквивалентность – это содержание одинакового активного вещества, который по результатам клинических исследований, обладает такой же эффективностью и безопасностью, как и препарат сравнения, с доказанными свойствами [8, с. 260-267; 9].
2. Основные этапы разработки генерических препаратов
Обязательным условием для утверждения на фармацевтическом рынке является биоэквивалентность генериков оригинальным препаратам. Однако между созданием оригинального препарата и его генерика существует значительная разница, в этапности процесса, его длительности и стоимости. Процесс создания оригинального лекарственного средства достаточно длительный и дорогостоящий, находящийся под жестким контролем и занимающий около 12–15 лет [7, с. 5-13; 12, с. 20-31]. Синтезированная молекула активного компонента подвергается доклиническим испытаниям безопасности и биологической активности по стандартам надлежащей лабораторной практики (GLP), клинические испытания на здоровых добровольцах и на пациентах по стандартам надлежащей клинической практики (GCP) – продолжается несколько лет [1, с. 87-94; 2, с. 23-26; 9; 10, с. 1991-2001; 11, с. 338-349]. Производство оригинального лекарственного препарата осуществляется в соответствии с международными стандартами надлежащей промышленной практики (GMP). Известно, что только 0,2% разработанных молекул доходит до рынка в виде лекарственного препарата. Очевидно, что главными преимущественными особенностями оригинальных лекарственных средств являются: доказанные эффективность и безопасность, инновационность и воспроизводимость эффекта [2, с. 23-26].
Процесс разработки и создания генерика происходит другим путем (рис.). Его разработка значительно отличается от такового оригинального препарата по большей степени масштабами исследований. Создание генерических препаратов начинается с поиска и анализа оригинального средства, аналог которого может расширить линейку препаратов с одинаковым действующим веществом. В обязательном порядке определяется наличие патентной защиты оригинала, в котором важное значение, имеют факторы защиты интеллектуальной собственности, в частности [1, с. 87-94; 2, с. 23-26; 3, с. 5-11; 9]:
- Базовый патент на вещество – защищает саму молекулу и ее синтез;
- Патент на готовую лекарственную форму – защищает состав препарата, определенное вспомогательное вещество для изменения биодоступности и др.
Необходимо отметить, что в отличие от коммерческого использования вещества, его исследование, разработка и производство не являются нарушением интеллектуальной собственности.
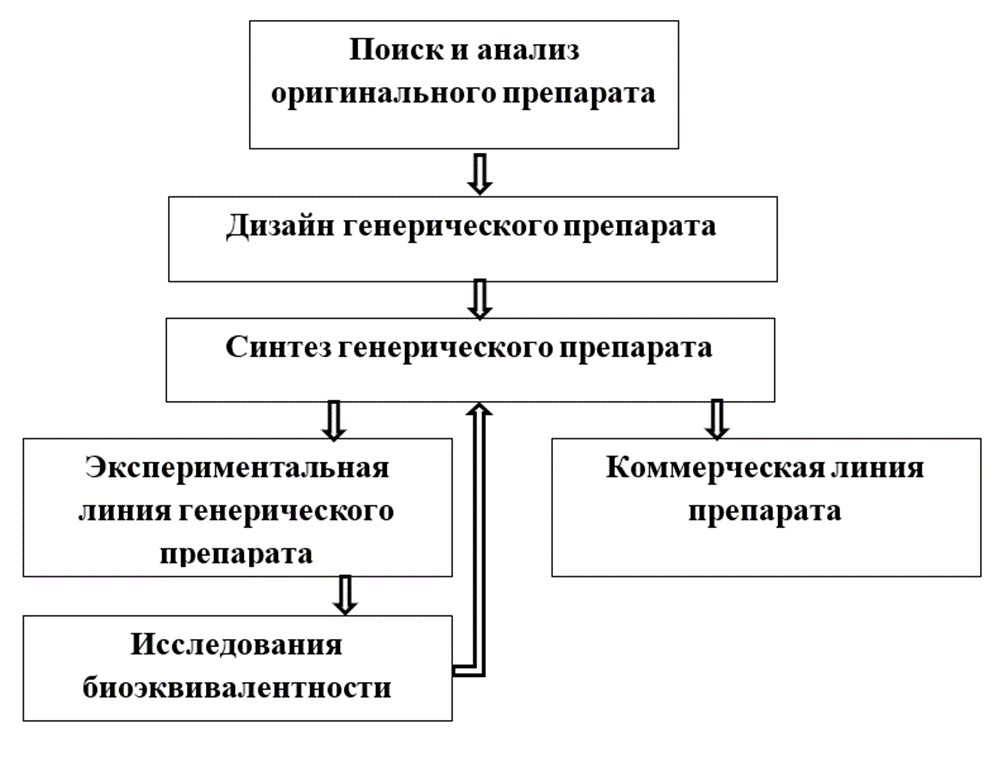
Рис. Схема разработки генерического лекарственного препарата
После нахождения подходящего оригинального вещества начинается разработка состава и технологии производства его аналога, после которых проводятся ограниченные клинические исследования. Для этого был предложен простой и недорогой процесс необходимый для утверждения генериковых версий оригинального препарата, который не требует многочисленных сравнительных доклинических или клинических исследований их безопасности и эффективности [12, с. 20-31]. После подтверждения их фармацевтической эквивалентности и биоэквивалентности по отношению к оригиналу автоматически предполагается и терапевтическая эквивалентность [10, с. 1991-2001; 11, с. 338-349; 12, с. 20-31]. Также, ответственным за фармацевтическую эквивалентность считается только основная активная субстанция без особого внимания к вспомогательным веществам, присутствующим во всех составах.
Заключительный этап включает процесс регистрации разработанного генерического вещества и занимает 3–12 месяцев [8, с. 260-267; 9; 10, с. 1991-2001]. При этом необходимо учитывать период эксклюзивности регистрационных данных препарата, поскольку регистрационное свидетельство на генерик может быть получено только после его истечения.
Поскольку при разработке генерических препаратов отсутствуют три этапа клинических исследований и перед регистрацией проводится только исследование биоэквивалентности, то себестоимости активной субстанции составляет не более 50%. В целом, разработка и регистрация генерического препарата занимает от 2 до 5 лет, в связи с чем, начинать разрабатывать этот вопрос, целесообразно за 4 года до окончания патентной защиты оригинального лекарственного средства [10, с. 1991-2001].
Теоретически возможно, что генерик может быть не соответствовать фармакокинетическим параметрам оригинала. Результатом может стать низкая эффективность или развитие нежелательных побочных реакций, связанных с передозировкой препарата. В связи с этим, как было отмечено выше, для всех генерических препаратов обязательно проводится исследование на биоэквивалентность. Сравнительное исследование должно проводиться по определенным правилам (GCP) и должно быть независимым, рандомизированным, контролируемым и длительным по времени и ограничиваться жесткими конечными точками [3, с. 5-11; 5, с. 1335-1355]. Для исследования фармакинетки привлекаются 20–30 клинически здоровых добровольцев, которые однократно принимают одну суточную дозу изучаемого генерика (для пролонгированного препарата – 5 суток приема); после мониторинга нескольких периодов полувыведения генерика, испытуемые добровольцы повторяют исследования с аналогичной дозой оригинального препарата. При анализе полученных данных различия двух графиков фармакокинетики не должны превышать 20% [4; 7, с. 5-13].
В последнее время одним из важных методов оценки непатентованных лекарственных препаратов являются количественные методы клинической фармакологии и моделирование (QMM), которые охватывают широкий спектр наборов инструментов, облегчающий процесс разработки и проверки непатентованных лекарственных средств, что может играть ключевую роль в модернизации оценки биоэквивалентности [11, с. 338-349; 12, с. 20-31]. Считается, что настоящий подход особенно перспективен для лекарственных препаратов местного действия и твердых пероральных форм.
3. Пути регулирования вопросов качества генерических препаратов в мировой фармацевтике
Потребность в эффективных и недорогих лекарствах определяет рост фармацевтического рынка, и соответственно, спрос на непатентованные лекарства. Важнейшими факторами, влияющими на качество непатентованных лекарств, являются чистота, эффективность, стабильность и скорость высвобождения, которые должны контролироваться в рамках соответствующих параметров для обеспечения желаемого качества лекарственного средства. Для получения сравнимого терапевтического эффекта генерики должны быть фармацевтическими, фармакокинетическими (биологическими) и терапевтическими эквивалентами оригинальному препарату [1, с. 87-94; 2, с. 23-26; 3, с. 5-11; 4; 5, с. 1335-1355; 8, с. 260-267].
Как рассматривалось выше, основным преимуществом генерика является его цена, которая на 30–80% дешевле, чем оригинальный препарат, что происходит за счет сокращения основных этапов его разработки. Однако, в целях снижения стоимости, многие фармацевтические компании ищут возможность приобретения наиболее дешевых субстанций или изменяют методы ее синтеза. Более того, качество генериков в значительной степени определяется качеством не только активной субстанции, но также зависит от вспомогательных веществ (наполнителей), т. к. они могут оказывать влияние на биодоступность, приводить к развитию токсических и аллергических реакций [11, с. 338-349; 12, с. 20-31]. Многие поставки субстанций происходят через большое количество посредников, сведения о месте производства обычно не афишируются, а готовый продукт рекламируется, как изготовленный в высокоразвитой стране. Таким образом, снижение стоимости генерических лекарственных препаратов порой достигается подходами, которые могут угрожать здоровью нации, и человечества в целом. К такому результату приводят приобретение некачественного первичного сырья, отсутствие клинических исследований, сравнительного анализа с оригиналом, изучения профиля безопасности.
Несоблюдение международных требований к генерикам влечет за собой огромные потери от «выгодной» цены вследствие затрат на повторные госпитализации, большие дозы лекарственных средств и лечение побочных эффектов. В связи с этим, требования к качеству безопасности генериков особенно высоки в европейских странах и США [2, с. 23-26; 8, с. 260-267; 9]. Разработка генерических препаратов в этой группе стран проходит под жестким контролем на каждом этапе пути от производителя к пациенту. Кроме этого, имеет место отрицательное отношение к биоэквивалентности, как к единственному способу оценки равнозначности лекарств, в связи с чем, обязательным является проведение клинических исследований на терапевтическую эквивалентность. Регистрация генериков длится в течение 1–3 лет до появления препарата на рынке и включает обязательную сертификацию GMP на все звенья производства и информирование о полном составе препарата, описание методов производства и контроля, результаты фармакологических тестов активной субстанции и конечного продукта.
Несмотря на контроль разработки генерических препаратов во многих развитых странах при замене оригинального препарата на аналог должно иметь веские причины [2, с. 23-26; 8, с. 260-267]. Это может объяснять тем, что биоэквивалентные торговые марки лекарств зачастую различаются по содержанию и качеству вспомогательных веществ, что может существенно изменить качество препарата, его биодоступность, привести к токсическим или аллергическим реакциям. Поэтому чаще всего генерические препараты применяются при социально значимых заболеваниях, имеющих высокую распространённость (артериальной гипертонии, хронической сердечной недостаточности, туберкулёзе, сахарном диабете и др.).
Немаловажным моментом является владение лечащего врача полноценной информацией о том, какой препарат является оригиналом, а также о качестве генерических препаратов [9; 10, с. 1991-2001]. В связи с этим, в некоторых европейских странах генерики разделены на группы «А» и «В». Код «А» присваивается препаратам, прошедшим клинические исследования на терапевтическую эквивалентность и имеющим отличия биоэквивалентности от оригинального лекарственного средства не более 5%. Генерики с кодом «А» могут являться в большинстве случаев полноценной заменой оригинальному препарату по финансовым соображениям. Генерикам, не прошедшим клинические испытания на терапевтическую эквивалентность присваивается код «В», в большинстве случаев он используется при заболеваниях, имеющих социальную значимость и широкую распространенность.
Доля воспроизведенных или генерических препаратов на фармацевтических рынках Российской Федерации составляет 78–95%, многие из которых имеют достаточный уровень качества, и в ходе исследований оценивается по тем же критериям, что и при регистрации в европейских странах (в соответствии со стандартом GMP) [1, с. 87-94; 2, с. 23-26; 6, с. 27-30] Многие препараты зарегистрированы в странах с развитой контрольно-разрешительной системой, к которым относятся страны – члены PIC (Конвенции о фармацевтических инспекциях) и PIC/S (Схемы сотрудничества фармацевтических инспекций): Также в России создана система Фармаконадзора, подразумевающая информирование Росздравнадзора о любых нежелательных побочных реакциях, которые может поступать как от врача, так и от пациента. На основе поступающих сообщений Росздравнадзор делает выводы о необходимости проверки лекарственного средства.
Заключение
Генерики являются необходимым звеном фармацевтического рынка, поскольку лечение только за счет оригинальных препаратов не может позволить себе ни одна страна в мире. Существенными преимуществами воспроизведенных препаратов являются ускоренный процесс разработки и относительно небольшие финансовые вложения, которые положительно влияют на регулирование цены для основной массы населения. Однако, снижение стоимости генерических лекарственных препаратов порой достигается подходами, как приобретение некачественного первичного сырья, отсутствие клинических исследований, сравнительного анализа с оригиналом, изучения профиля безопасности. Несоблюдение международных требований к воспроизведенным препаратам влечет за собой огромные потери от «выгодной» цены вследствие затрат на повторные госпитализации, и лечение побочных эффектов. В связи, с этим необходимым условием для сохранения здоровья нации является разработка качественных аналогов оригинального препарата, что достигается путем соблюдения международных требований к генерикам и ужесточения качества за их производством.
